Ma la talassemia è una malattia sola o ci sono tante forme della stessa?
In effetti non si dovrebbe parlare in maniera generica di talassemia perché si possono distinguere diverse forme sulla base del difetto genetico che determina la perdita di emoglobina e di conseguenza una sintomatologia più o meno grave.
L’alfa-talassemia è causata da difetti del gene della catena alfa dell’emoglobina (una porzione della proteina), con quadri di gravità variabile, da forme silenti a forme mortali come il cosiddetto idrope fetale di Bart, con formazione di emoglobina anomala nel feto. In genere il decesso avviene quasi sempre in utero o poco dopo la nascita.
La beta-talassemia è la forma più diffusa in Italia ed è dovuta alla riduzione o assenza della catena beta dell’emoglobina. La beta-talassemia minor è di gran lunga la più frequente ed è caratterizzata da sintomi lievi (spesso addirittura assenti) che non richiedono alcun trattamento farmacologico. Ma c’è una forma più grave, la talassemia major, chiamata anche malattia di Cooley, una condizione patologica che insorge nel neonato con i classici sintomi di una grave anemia (debolezza, pallore, ittero, ritardo della crescita).
Se non vengono trattati, questi sintomi tendono ad aggravarsi portando a ulteriori alterazioni come le deformità scheletriche (deformazione delle ossa del volto e delle ossa lunghe di braccia e gambe). La causa principale delle complicazioni è dovuta al midollo osseo che, per compensare l’ipossia cronica, viene stimolato, da un ormone chiamato eritropoietina, ad aumentare la produzione di globuli rossi (eritrociti). Questi pazienti mostrano anche gonfiore addominale per l’ingrossamento di fegato e milza, che reagiscono allo stesso modo alle conseguenze dell’ipossia.
Esiste, infine, una forma di talassemia intermedia con sintomatologia e gravità variabile.
Cosa si può fare in queste condizioni così disperate?
L’unica terapia valida per questi pazienti sono delle continue trasfusioni di sangue. Si crea così una dipendenza trasfusionale che, da un lato consente una crescita e uno sviluppo normale del bambino, ma dall’altro ha delle importanti conseguenze sulla qualità e aspettativa di vita.
Le trasfusioni, infatti, portano ad un eccessivo accumulo di ferro con danni a diversi organi tra cui cuore, fegato e ghiandole endocrine. Per evitare il sovraccarico di ferro, a questi pazienti vengono somministrati farmaci chelanti (deferoxamina, deferiprone, deferasirox) con lo scopo di rimuovere l’eccesso di ferro, ma spesso con risultati non soddisfacenti.
Esistono terapie in grado di curare in maniera definitiva la talassemia?
Si, esistono e sono in fase di evoluzione grazie alle migliori conoscenze della patologia anche se fino a poco tempo fa l’unico trattamento curativo era rappresentato dal trapianto di midollo osseo. Cioè la sostituzione delle cellule del sangue malate con quelle sane di un donatore compatibile.
Grazie al trapianto, il paziente riceve le cellule (chiamate cellule staminali ematopoietiche) che poi producono i globuli rossi sani capaci di trasportare l’ossigeno ai tessuti.
Tuttavia, questa metodica presenta numerosi rischi. C’è necessità di usare farmaci chemioterapici ad alte dosi per favorire il trapianto (condizionamento pre-trapianto). Questo trattamento, che serve a eliminare tutte le cellule malate, causa anche la riduzione delle cellule immunitarie, con la conseguenza di mettere il soggetto a rischio di contrarre infezioni gravi, e delle piastrine, con rischio di emorragie anche fatali.
Inoltre, con il trapianto, il paziente può sviluppare la cosiddetta malattia del trapianto contro l’ospite (graft versus host disease, GvHD). Questa consiste nella reazione delle cellule ricevute con il trapianto contro quelle del paziente (di organi e tessuti), che non riconoscono come proprie. Per evitare questo fenomeno, ma anche il rigetto del midollo del donatore, è necessario usare farmaci immunosoppressori. Inoltre, non è sempre possibile trovare un donatore compatibile.
C’è quindi anche altro per evitare le complicazioni del trapianto di midollo osseo?
Si, gli scienziati hanno studiato la possibilità di trovare terapie farmacologiche efficaci e sicure come alternative valide al trapianto. Ecco che, molto recentemente (2020), l’Agenzia Europea dei Medicinali (EMA) ha autorizzato una nuova terapia per pazienti adulti con anemia trasfusione-dipendente, associata a beta-talassemia.
Si tratta del luspatercept, una proteina prodotta in laboratorio con la tecnologia del DNA ricombinante che promuove nei pazienti la corretta maturazione dei globuli rossi, riducendo di almeno un terzo il numero di trasfusioni. Pur essendo approvato in Europa, in Italia il farmaco non ha ancora ricevuto l’autorizzazione dall’Agenzia Italiana del Farmaco (AIFA). Da noi è comunque possibile utilizzarlo nell’ambito di un programma di uso compassionevole fornito dall’Azienda produttrice, secondo criteri predefiniti.
Oggi si pensa alla terapia genica. Quali sono le prove a favore di questa nuova proposta terapeutica salvavita?
La terapia genica ha l’obiettivo di fornire una cura definitiva, fornendo all’organismo una copia corretta del gene difettoso o un altro gene che possa compensarne il malfunzionamento.
Una modalità per trasferire un gene corretto nel paziente consiste nel prelievo delle cellule malate del paziente. Queste, portate in laboratorio, vengono modificate con l’introduzione del gene corretto e quindi re-introdotte nel paziente. Anche in questo caso è necessario rimuovere tutte le cellule malate, prima della re-introduzione delle cellule con il gene corretto, e questo si fa con la chemioterapia come descritto in precedenza.
Nel 2019, EMA ha autorizzato la prima terapia genica per una forma di beta-talassemia, in cui la proteina beta non sia completamente assente, in bambini (di almeno 12 anni) e in adulti candidabili al trapianto di midollo ma che non abbiano un donatore compatibile. Il trattamento è denominato Zynteglo®. Anche in questo caso, si prelevano le cellule staminali ematopoietiche del paziente, si modificano in laboratorio con l’aggiunta del gene della beta-emoglobina.
Una volta re-infuse, le cellule modificate vanno a ripopolare il midollo osseo e iniziano a produrre le cellule del sangue. Si tratta di un trattamento personalizzato, in cui il ‘farmaco’ è costituito dalle cellule dello stesso paziente e può essere somministrato esclusivamente al paziente da cui sono state prelevate.
Negli studi clinici effettuati, il trattamento ha dimostrato di poter risolvere l’anemia dopo una unica somministrazione delle cellule geneticamente modificate, mantenendo l’attività potenzialmente per tutta la vita e riducendo o azzerando la necessità di ricevere trasfusioni. I principali effetti collaterali sono stati i ridotti livelli di piastrine (con il rischio di emorragie) e quelli dalla terapia di condizionamento con i chemioterapici. Non sono ancora noti gli effetti a lungo termine e sono in corso studi aggiuntivi per valutarli.
Zynteglo® ha ottenuto una “autorizzazione subordinata a condizioni”. Ciò significa che in futuro l’azienda farmaceutica dovrà fornire ulteriori informazioni su questo farmaco. EMA esaminerà le nuove informazioni disponibili e darà disposizioni conseguenti sull’utilizzo.
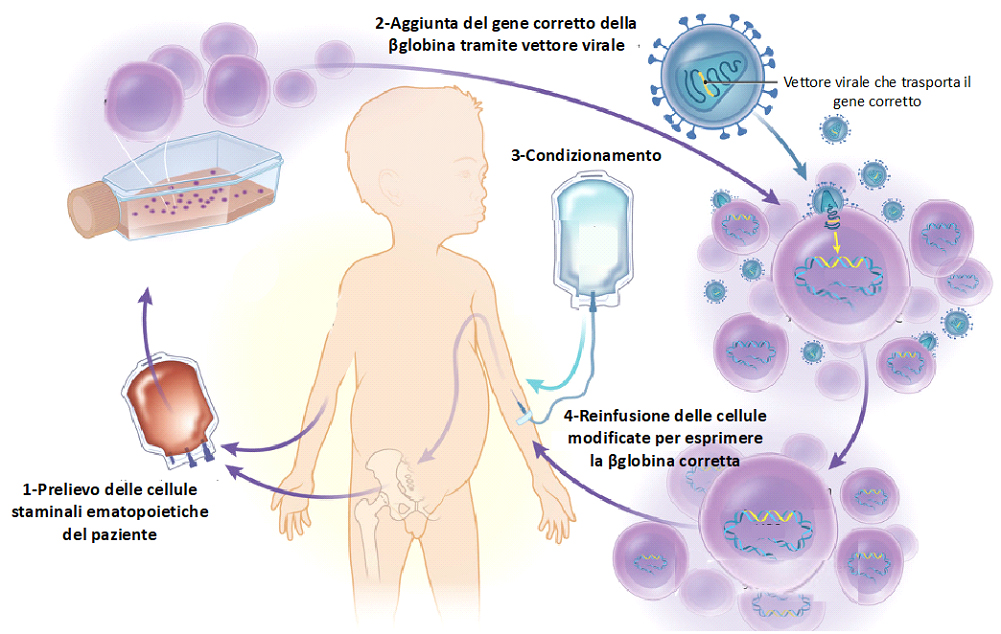
Figura 1 Terapia genica per la beta-talassemia (Modificata da KA High, MG Roncarolo. N Engl J Med 2019;381:455-464.)
Ci sono anche altre strategie terapeutiche in studio per la talassemia?
Sono in corso numerosi studi su nuove strategie terapeutiche. Ad esempio, c’è una terapia che si basa sulla tecnica dell’editing genetico con lo scopo di far esprimere il gene della gamma-globina tipica dell’emoglobina fetale (HbF), per compensare la carenza delle catene beta tipiche dell’emoglobina adulta.
La prima somministrazione in Italia di questa terapia chiamata CTX001, basata sulla tecnica di editing genetico, è stata effettuata il 17 novembre 2020, dopo che i primi risultati di uno studio clinico internazionale (CLIMB-111) avevano dato riscontri positivi.
Il trattamento si basa su un processo di manipolazione genetica di cellule staminali ematopoietiche prelevate dai pazienti allo scopo di riprogrammarle per produrre alti livelli di HbF nei globuli rossi. La HbF è una forma di emoglobina presente alla nascita, che viene in seguito sostituita dalla sua forma adulta. L'aumento dei livelli di HbF, dopo somministrazione di CTX001, potrebbe portare a una significativa riduzione se non all’azzeramento del numero di trasfusioni.
Resta comunque valida l’importanza di uno screening genetico dei portatori delle mutazioni responsabili della talassemia per una adeguata prevenzione e gestione della patologia.
Riferimenti bibliografici e sitografici:
⦁ Cappellini MD at al. A Phase 3 Trial of Luspatercept in Patients with Transfusion-Dependent β-Thalassemia. N Engl J Med. 2020 Mar 26;382(13):1219-1231
⦁ Frangoul H et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N Engl J Med. 2021 Jan 21;384(3):252-260
⦁ Rund D, Rachmilewitz E. Beta-thalassemia. N Engl J Med. 2005 Sep 15;353(11):1135-46
⦁ Thompson AA et al. Gene Therapy in Patients with Transfusion-Dependent β-Thalassemia. N Engl J Med. 2018 Apr 19;378(16):1479-1493. doi: 10.1056/NEJMoa1705342. PMID: 29669226.
⦁ Harrison C. First gene therapy for beta-thalassemia approved. Nat Biotechnol. 2019 Oct;37(10):1102-1103. doi: 10.1038/d41587-019-00026-3.